美國醫生首次治療了子宮內患有快速進展先天性疾病的胎兒。
而不是給患有嚴重神經肌肉並發症的孩子用藥後他們出生後,一項新的案例研究詳細介紹了一位準母親在懷孕期間如何同意為正在發育的胎兒服用這種藥物。
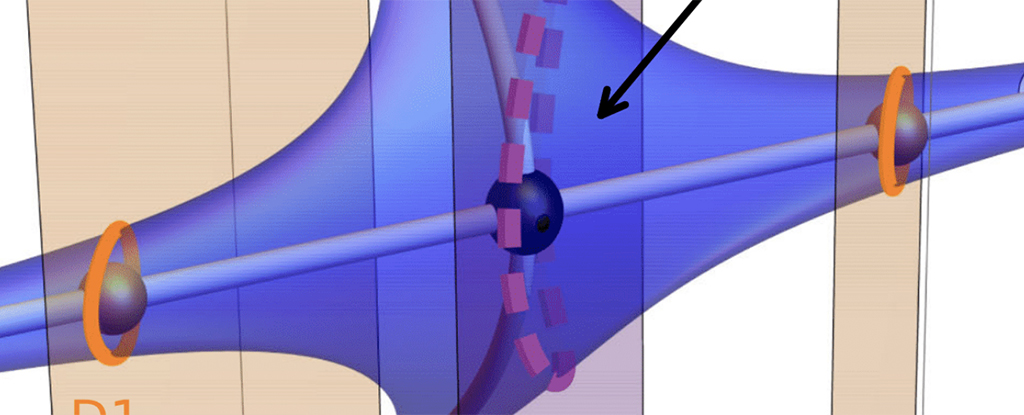
產前測試顯示,她的胎兒攜帶兩種基因突變,表明 1 型脊髓性肌萎縮症 (SMA),通常會導致嚴重的肌肉無力和呼吸困難出生後六個月內。
從歷史上看,大多數患有 1 型 SMA 的兒童都會死亡到他們兩歲生日時,通常是由於呼吸衰竭。然而,這項研究中的孩子到目前為止已經兩歲半了,沒有任何症狀跡象。
接受治療的孩子的父母之前曾因脊髓性肌萎縮症而失去孩子,當測試顯示他們最近的胎兒也患有進行性神經肌肉疾病時,他們想知道是否可以更早開始治療。
對於這一個案,FDA 批准了口服藥物 risdiplam 的早期給藥,其商品名為 Evrysdi,由製藥公司 F. Hoffmann-La Roche AG 擁有。
多年來,隨機對照試驗研究表明,risdiplam 在治療新生兒脊髓性肌萎縮症方面既安全又有效,而且孩子開始接受藥物治療時年齡越小,總體效果就越好。
在這項新研究中,懷孕的母親在分娩前連續六週每天服用一劑 risdiplam。測試發現,藥物直接滴入她的臍帶血和沐浴胎兒的羊水中。出生後,每天口服藥物的是嬰兒,而不是母親。
小兒神經科醫生 Michelle Farrar 正在研究一種微創手術基因療法治療對於澳大利亞的 SMA,告訴斯姆里蒂·馬拉帕蒂自然患有 SMA 的女嬰“已得到有效治療,即使在出生後 30 個月也沒有出現任何症狀”。
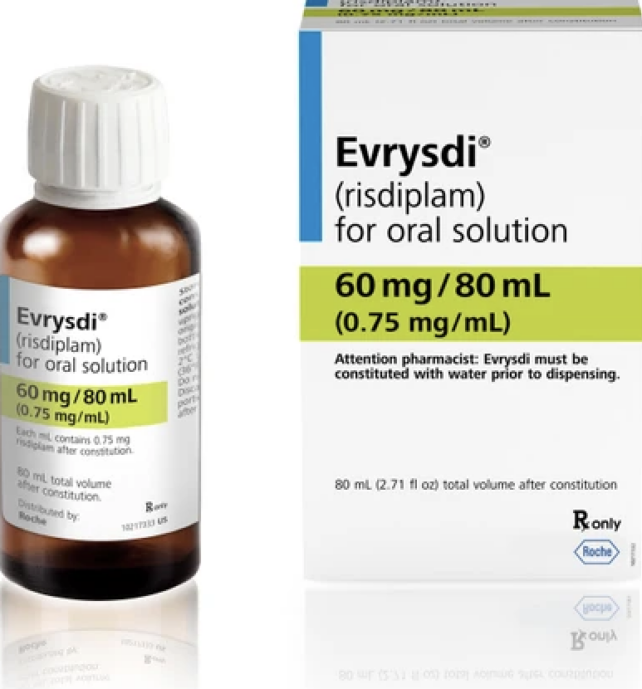
2020年,risdiplam首次得到正式認可的美國食品和藥物管理局 (FDA) 針對患有脊髓性肌萎縮症的兒童推出了這種藥物,但僅限兩個月以上的兒童。
最近的臨床試驗然而,由羅氏資助的發現大多數兒童使用 risdiplam 治療脊髓性肌萎縮症前經過兩年的治療後,六周大的孩子可以自己吞嚥、進食、坐下、站立和行走。不需要永久通風。
Risdiplam 通過增加一種稱為運動神經元存活 (SMN) 蛋白的關鍵蛋白質的濃度來防止大腦和身體神經細胞的退化。那些具有導致脊髓性肌萎縮症的基因突變的人缺乏活性 SMN 蛋白。
“對於患有 SMA 的兒童,運動神經元變性在症狀出現之前就開始了,因此如果我們希望保持肌肉功能,時間至關重要,”說兒科神經學家 Laurent Servais 在羅氏的新聞稿中2024年。
“令人振奮的是,通過 Evrysdi 的早期干預,這些孩子已經實現了坐、站和行走等重要的里程碑,而如果不進行治療,這些通常是無法實現的。”
美國最近的案例研究是否會激勵和鼓勵醫生和家長在早期開發階段試用 risdiplam,還有待觀察。
“這個單一病例的結果不能一概而論,但可能支持考慮對子宮內發現的 SMA 進行產前 risdiplam 治療,”寫信件的作者。
該研究發表於新英格蘭醫學雜誌。